
Epigentic Studies
2. Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations
Authors: Paul F. Gugger, Sorel Fitz-Gibbon, Matteo Pellegrini, and Victoria L. Sork
Molecular Ecology, in press
Abstract
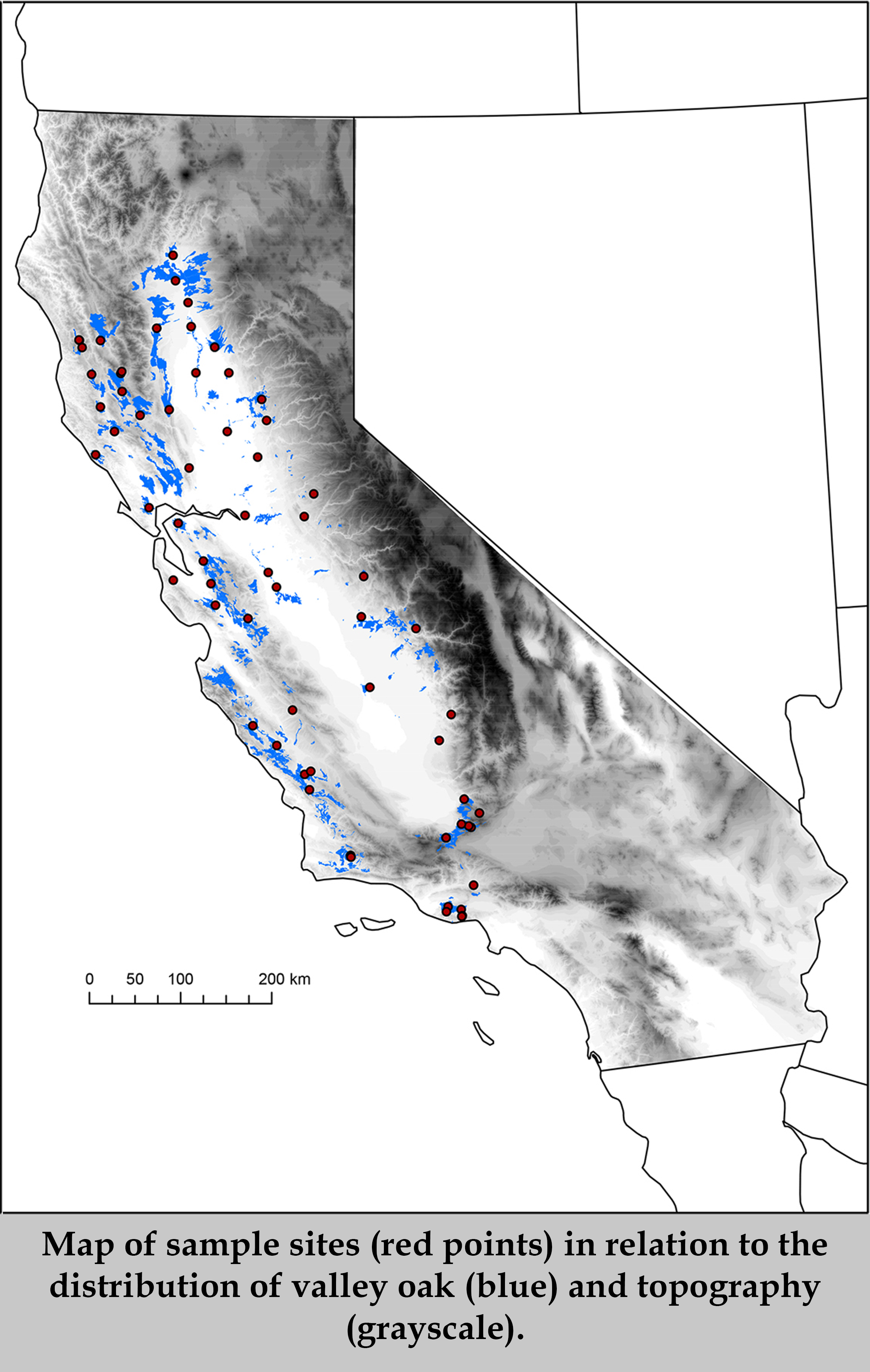
DNA methylation in plants affects transposon silencing, transcriptional regulation, and thus phenotypic variation. One unanswered question is whether DNA methylation could be involved in local adaptation of plant populations to their environments. If methylation alters phenotypes to improve plant response to the environment, then methylation sites or the genes that affect them could be a target of natural selection. Using reduced-representation bisulfite sequencing (RRBS) data, we assessed whether climate is associated with variation in DNA methylation levels among 58 naturally occurring, species-wide samples of valley oak (Quercus lobata) collected across climate gradients. We identified the genomic context of these variants referencing a new draft valley oak genome sequence. Methylation data were obtained for 341,107 cytosines, of which we deemed 57,488 as single-methylation variants (SMVs), found in the CG, CHG, and CHH sequence contexts. Environmental association analyses revealed 43 specific SMVs that are significantly associated with any of four climate variables, the majority of which are associated with mean maximum temperature. The 43 climate-associated SMVs tend to occur in or near genes, several of which have known involvement in plant response to environment. Multivariate analyses show that climate and spatial variables explain more overall variance in CG-SMVs among individuals than in SNPs, CHG-SMVs, or CHH-SMVs. Together, these results from natural oak populations provide initial evidence for a role of CG methylation in locally adaptive evolution or plasticity in plant response.
Data links for this study:
--Valley Oak Genome 0.5 (reduced) FASTA file download
-Valley Oak Genome 0.5 subset annotation gff file download
-SNPs, SMVs, and BAM files at Dryad
Authors: Alexander Platt, Paul F. Gugger, Matteo Pellegrini, and Victoria L. Sork
Molecular Ecology
Abstract
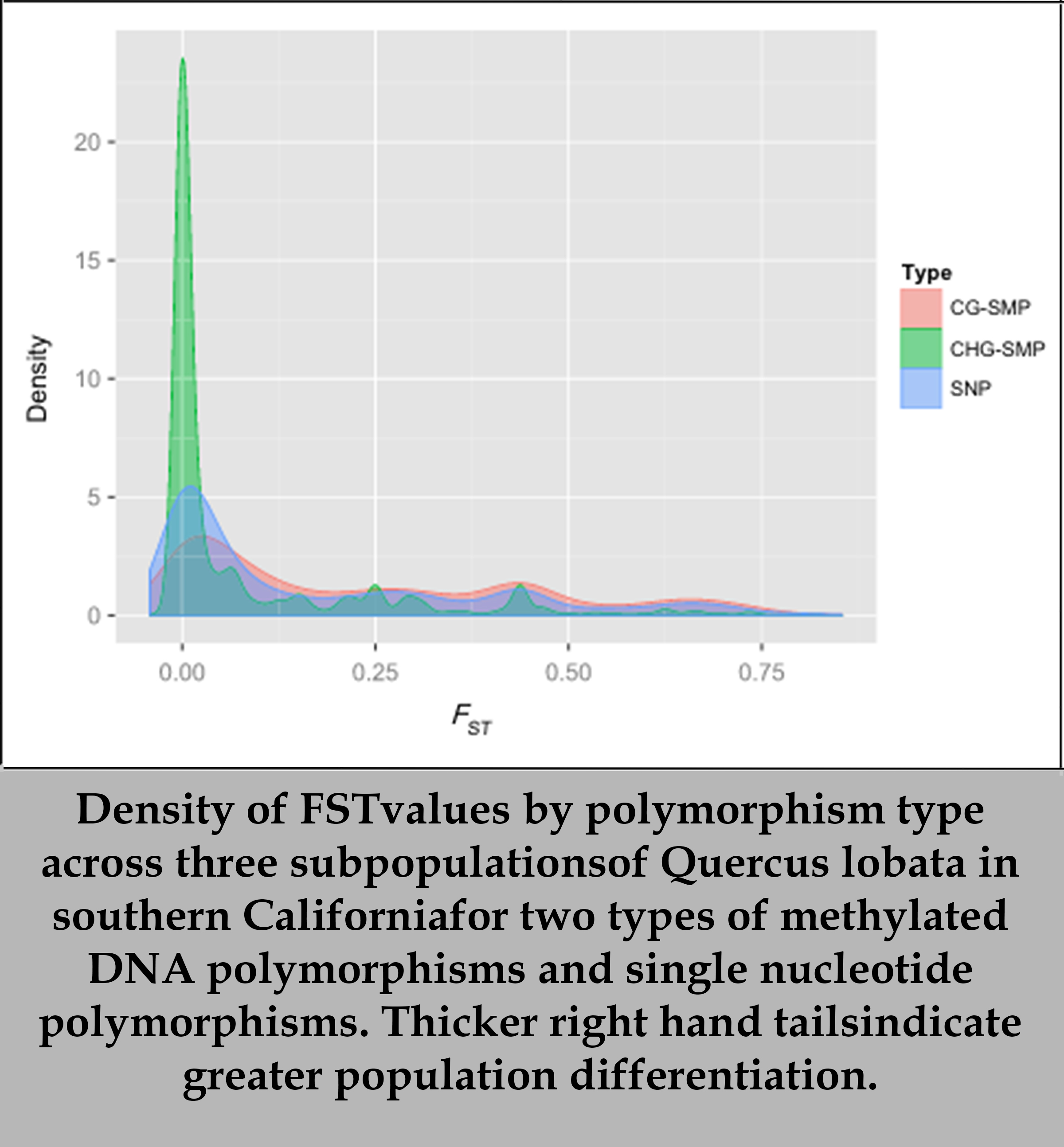
It has long been known that adaptive evolution can occur through genetic mutations in DNA sequence, but it is unclear whether adaptive evolution can occur through analogous epigenetic mechanisms, such as through DNA methylation. If epigenetic variation contributes directly to evolution, species under threat of disease, invasive competition, climate change or other stresses would have greater stores of variation from which to draw. We looked for evidence of natural selection acting on variably methylated DNA sites using population genomic analysis across three climatologically distinct populations of valley oaks. We found patterns of genetic and epigenetic differentiations that indicate local adaptation is operating on large portions of the oak genome. While CHG methyl polymorphisms are not playing a significant role and would make poor targets for natural selection, our findings suggest that CpG methyl polymor-phisms as a whole are involved in local adaptation, either directly or through linkage to regions under selection.
Provenance Study
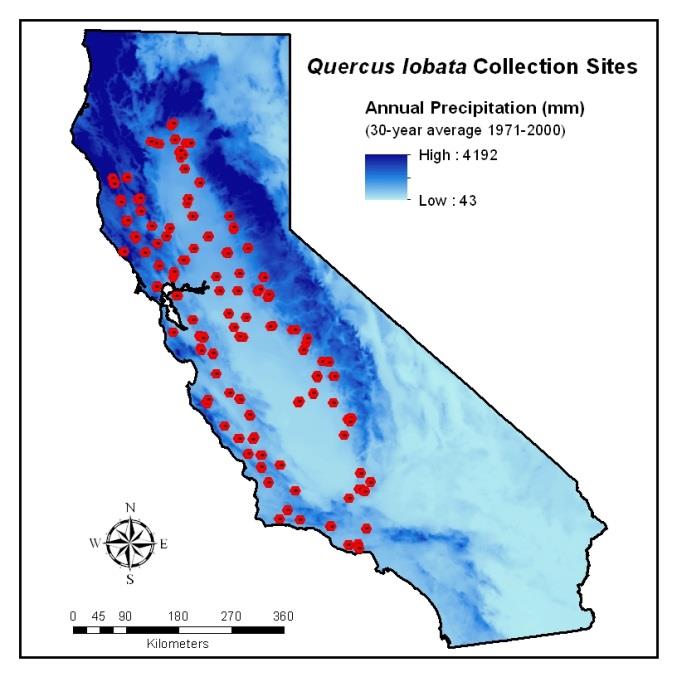
Our sequenced genome for valley oak is a tool that will allow us to examine closely the relationship between genetic variation across the entire genome, and observed traits from trees in the field. However, trait values are strongly associated with the environment where a tree is found. Therefore, traits need to be evaluated in a common garden setting. In 2012, we collected acorns from 694 maternal trees from 95 different collecting sites across the range of the species (see map below). The seedlings resulting from these acorns were transplanted into two field sites in the winter of 2014/15.
We are collecting trait data on these seedlings, including survival and growth. In addition, we have been examining the phenology of bud burst, and recording when trees start to grow in the spring. Using our reference genome, we plan to collect genomic data from the parental trees, and relate the traits we measured in a the common garden with variation across the entire genome. In this way, we’ll be able to identify SNPs, genes and alleles associated with the observed traits.
All growth data from the provenance test will be available publicly at the Center for Forest Provenance Data found here as well as the USFS R&D data archive.
Publications:
1) Delfino-Mix A, JW, PF Gugger, C Liang, VL Sork. In press. Establishing a range-wide provenance test in valley oak (Quercus lobata Née) at two California sites. Proceedings of the 7th California Oak Symposium: Managing Oak Woodlands in a Dynamic World. Visalia, CA. PDF link
2) Albarrán-Lara AL, JW Wright, PF Gugger, A Delfino-Mix, JM Peñaloza Ramírez, VL Sork. In press. Phenotypic variation in California populations of valley oak (Quercus lobata Née) along three elevation transects. Proceedings of the 7th California Oak Symposium: Managing Oak Woodlands in a Dynamic World. Visalia, CA. PDF link



Images on the right side of the page show bud development.





































