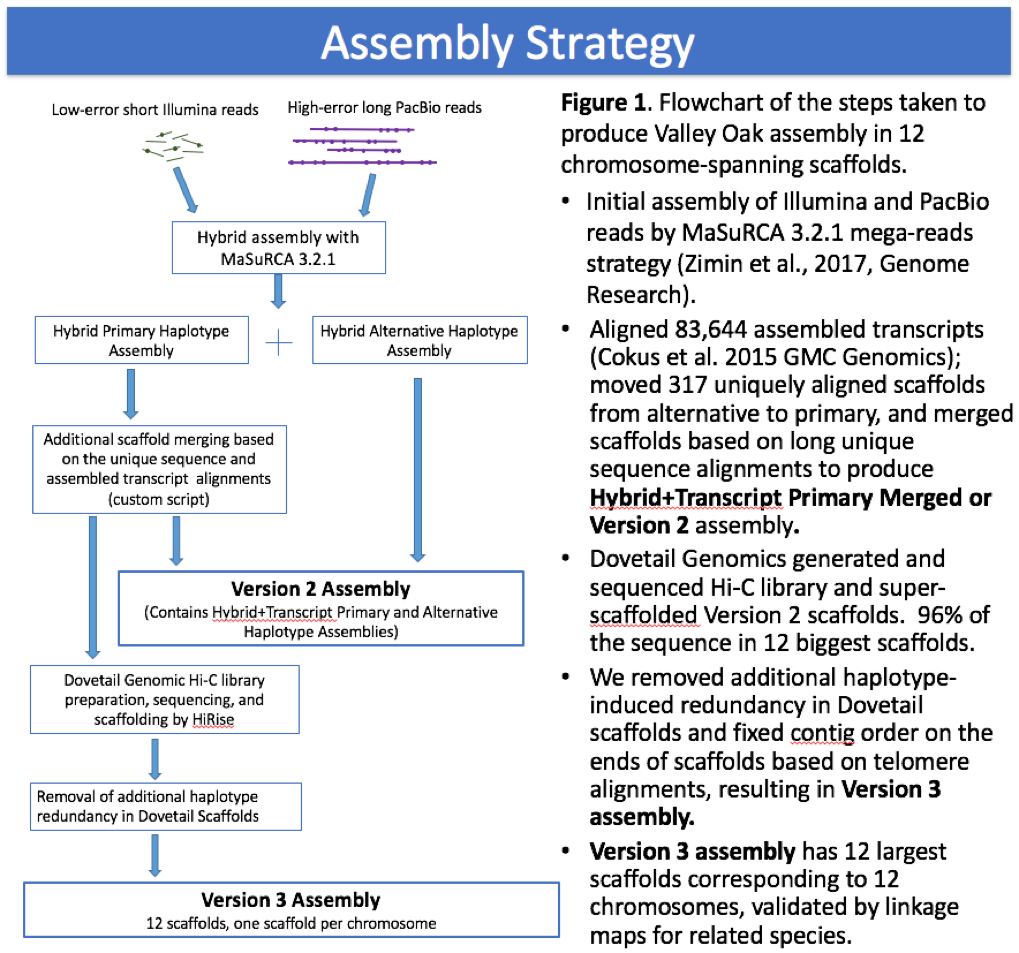
Valley Oak Genome v 3.0
 This genome is our final assembly for Quercus lobata SW786. It includes 96% of the sequence in twelve chromosome length scaffolds labeled chr1, chr2,.. chr12. There are an additional 2,016 unplaced scaffolds ordered by size and ranging from 1411,17 to 1001 bps. The gene models are also available below, as well as other genomic resources.
This genome is our final assembly for Quercus lobata SW786. It includes 96% of the sequence in twelve chromosome length scaffolds labeled chr1, chr2,.. chr12. There are an additional 2,016 unplaced scaffolds ordered by size and ranging from 1411,17 to 1001 bps. The gene models are also available below, as well as other genomic resources.
The manuscript is in prep:
V. L. Sork, S. J. Cokus, S. T. Fitz-Gibbon, A. Zimin, D. Puiu, J. A. Garcia, P. F. Gugger, C.L. Henriquez, Y. Xhen, K. E. Lohmueller, M. Pellegrini, S. L. Salzberg. 2021. High-quality genome and methylomes illustrate features underlying evolutionary success of oaks
In Revision.
This manuscript and website reports and analyzes the valley oak version 3.0 assembly. A small number of contaminants (found exclusively among non-chromosomal scaffolds) were subsequently identified and deleted, leading to the current (at time of publication) version 3.2 appearing at NCBI. These minor deletions do not significantly change our analyses or their interpretations.
You are welcome to use the genome and other resources. When using valley oak genome v3.0+ and associated annotation, please cite the preprint if the above manuscript has not yet been published: https://www.biorxiv.org/content/10.1101/2021.04.09.439191v1
Link to NCBI Umbrella BioProject —PRJNA781973
Download
Please note, some of the below analysis files were run on a version of the genome prior to deleting 18 scaffolds (identified to be derived from organelles) and replacing them with assembled versions: chrC, chrM1, chrM2 & chrM3. See details below under “Organelle Contigs”. Additionally 381,173 bps were converted to `N’s after being identified as inserted mis-assembly of mitochrondrial sequence: inclusive-inclusive range chr1:[+]29726880..30108053.
Assembled Genome
GZIP’d Final Assembly, including 12 chromosomes, 2016 unplaced scaffolds, a complete chloroplast contig, and 3 mitochondrial contigs (~240MB) — Qlobata.v3.0.RptMsk4.0.6.on-RptMdl1.0.8.softmasked.fasta.gz
– RepeatMasker intervals and all Ns are lower case in the above file
GZIP’d Alternate Contigs (from genomic regions where the two haplotypes assembled separately) (~117MB) — Qlobata.v3.0.alternateContigs.fasta.gz
Alternate Contigs extra information, sizes and relative coverage (~0.5MB) — Qlobata.v3.0.alternateContigs.info.txt
Protein Coding Gene Models, various formats
GZIP’d Gene Models, gtf format (~6.3MB) — Qlobata.v3.0.PCG.gtf.gz
GZIP’d Gene Models, bed12 format (~2MB) — Qlobata.v3.PCG.bed.gz
GZIP’d Coding sequences (~13MB) — Qlobata.v3.0.PCG.CDS.fasta.gz
GZIP’d Protein sequences (~8MB) — Qlobata.v3.0.PCG.prot.fasta.gz
Protein Functional Names, via PANTHER (~2MB) Oak-NAMING-GENES.FIN1-viaPANTHER–V0-20200626.txt
If you download the genome sequence from NCBI, you may prefer this version of the gene model annotation, using NCBI contig names for chromosomes 1-12 (~6.3MB) — Qlobata.v3.0.PCG.NCBIchromosomeNames.gtf.gz
Gene Functions – InterProScan 5.34-73.0 run on 39,373 protein-coding gene models
GZIP’s TAR bundle of all IPS files listed below (~467 MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0–BUNDLE.tar.gz
– OR –
GZIP’d IPS GFF3 format (~17MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.gff3.gz
GZIP’d FASTA file with sequence fragments referred to from the GFF3 file (~15MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.gff3fasta.gz
GZIP’d IPS main tab-separated format (~10MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.tsv.gz
GZIP’d IPS active sites tab-separated format (~3MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.sites.tsv.gz
GZIP’d IPS XML format (~49MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.xml.gz
GZIP’d IPS JSON format (~49MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.json.gz
GZIP’d TAR archive of IPS HTML outputs per gene (~188MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.HTMLs.tar.gz
GZIP’d TAR archive of IPS SVG drawing per gene (~156MB) — Qlobata.v3.0.PCG.InterProScan5.34-73.0.SVGs.tar.gz
Orthologs – OMA Stand-alone version 2.3.1 run on Arabdopsis, Q. lobata, Q. robur & Q. suber
`ARATH’ (Arabidopsis thaliana: Ensembl Plants 38 TAIR10), `QueLo’ Quercus lobata (39,373 PCGs matching Sork et al. 2020 Figure 4A), `QueRo’ Quercus robur (25,808 PCGs matching Sork et al. 2020 Figure 4A), `QueSu’ Quercus suber (49,388 PCGs matching Sork et al. 2020 Figure 4A).
GZIP’s TAR (~45MB) — OMA2.3.1–ARATH-QueLo-QueRo-QueSu–BUNDLE.tar.gz
Repeats – RepeatModeler open-1.0.8 & RepeatMasker open-4.0.6
GZIP’d TAR archive of Repeat Modeler, full output (~573MB) — Qlobata.v3.0.RepeatModeler-open-1.0.8–BUNDLE.tar.gz
GZIP’d FASTA Repeat Modeler, consensi, passed to Repeat Masker (~0.5MB) — Qlobata.v3.0.RepeatModeler-open-1.0.8.consensi.fa.classified.gz
GZIP’d TAR archive of Repeat Masker full output, using above consensi file (~1.3GB) — Qlobata.v3.0.RptMsk4.0.6.on-RptMdl1.0.8–BUNDLE.tar.gz
GZIP’d UCSC BED6-format, Repeat Masker Repeat Families (~20MB) — Qlobata.v3.0.RptMsk4.0.6.on-RptMdl1.0.8.bed.gz
– the BED score column is 10 times the GFF file scores
– upper/lowercase of `RF:’/`rf:’ in BED column 4 follows as determined by RepeatMasker
GZIP’d list of Repeat Families combined to make each Super Family (~43KB) — Qlobata.v3.0.RptMsk4.0.6.on-RptMdl1.0.8.SFtoMemberFams.txt.gz
GZIP’d UCSC BED4-format Super Families (~14MB) — Qlobata.v3.0.RptMsk4.0.6.on-RptMdl1.0.8.SF.bed.gz
Repeats – LTR Harvest & LTR Digest from GenomeTools v.1.5.9
GZIP’s GFF listing LTR transposable elements — Qlobata.v3.0.QLz.LTRharvest-LTRdigest-1.5.9.gff.gz
Alignments and 2-D visualizations
Nucleotide- and amino acid-level alignments and 2-D visualizations of sequence similarity, Ks, and shared Pfam domains, variously within and between genomes for Q. lobata, Q. robur, Q. suber, Populus, Eucalyptus, Theobroma, and Coffea available here
RNAseq
Fastq and summary wig files for SW786 bud, leaf & stem RNA-seq are available at NCBI GEO: GSE174828
PacBio RNA Sequencing (IsoSeq)
RNA long read sequencing: raw bam files for SW786 bud, leaf & stem are available at NCBI GEO: GSE174827
Methylation – whole genome bisulfite sequencing
WGBS fastq, bwa-meth bam and methylpy files for SW786 bud, catkin & youngleaf are available at NCBI GEO: GSE174826
methylpy output also here… (see https://pypi.org/project/methylpy/)
GZIP’d methylpy output, bud CG (~MB) — allc_bud.CG.tsv.gz
GZIP’d methylpy output, bud CHG (~MB) — allc_bud.CHG.tsv.gz
GZIP’d methylpy output, bud CHH (~MB) — allc_bud.CHH.tsv.gz
GZIP’d methylpy output, catkin CG (~MB) — allc_catkin.CG.tsv.gz
GZIP’d methylpy output, catkin CHG (~MB) — allc_catkin.CHG.tsv.gz
GZIP’d methylpy output, catkin CHH (~MB) — allc_catkin.CHH.tsv.gz
GZIP’d methylpy output, youngLeaf CG (~MB) — allc_youngLeaf.CG.tsv.gz
GZIP’d methylpy output, youngLeaf CHG (~MB) — allc_youngLeaf.CHG.tsv.gz
GZIP’d methylpy output, youngLeaf CHH (~MB) — allc_youngLeaf.CHH.tsv.gz
PSMC_inference, wgs for 19 California Q. lobata samples
fastq files available at NCBI: PRJNA729978 (also includes many other Q. lobata samples)
filtered variants, scripts, masks, PSMC inputs and outputs for reference genomes and resequenced genomes are available here (github) and here (box).
Organelle Contigs
Some of the above analysis files were run on a version of the genome prior to deleting 18 scaffolds deemed organelle derived and replacing them with the following contigs. These organelle contigs, ChrC, ChrM1, ChrM2 & ChrM3, are also included in the full genome download: Qlobata.v3.0.fasta.gz
GZIP’d FASTA chloroplast (1 contig, 161,289 bp) — Qlobata.v3.0.chloroplast.fasta.gz
GZIP’d GFF chloroplast (~3KB) — Qlobata.v3.0.chloroplast.gff.gz
GZIP’d FASTA mitochondrion (3 contigs, 444,512 bp) — Qlobata.v3.0.mitochondrion.fasta.gz
Scaffolds removed: Scq3eQI_83 Scq3eQI_674 Scq3eQI_14 Scq3eQI_18 Scq3eQI_787 Scq3eQI_1688 Scq3eQI_1766 Scq3eQI_789 Scq3eQI_1489 Scq3eQI_1288 Scq3eQI_972 Scq3eQI_771 Scq3eQI_795 Scq3eQI_672 Scq3eQI_1050 Scq3eQI_978 Scq3eQI_839 Scq3eQI_997)
Browser view of genomic resources of Q. lobata
Link to live browser will be posted by early July 2020.
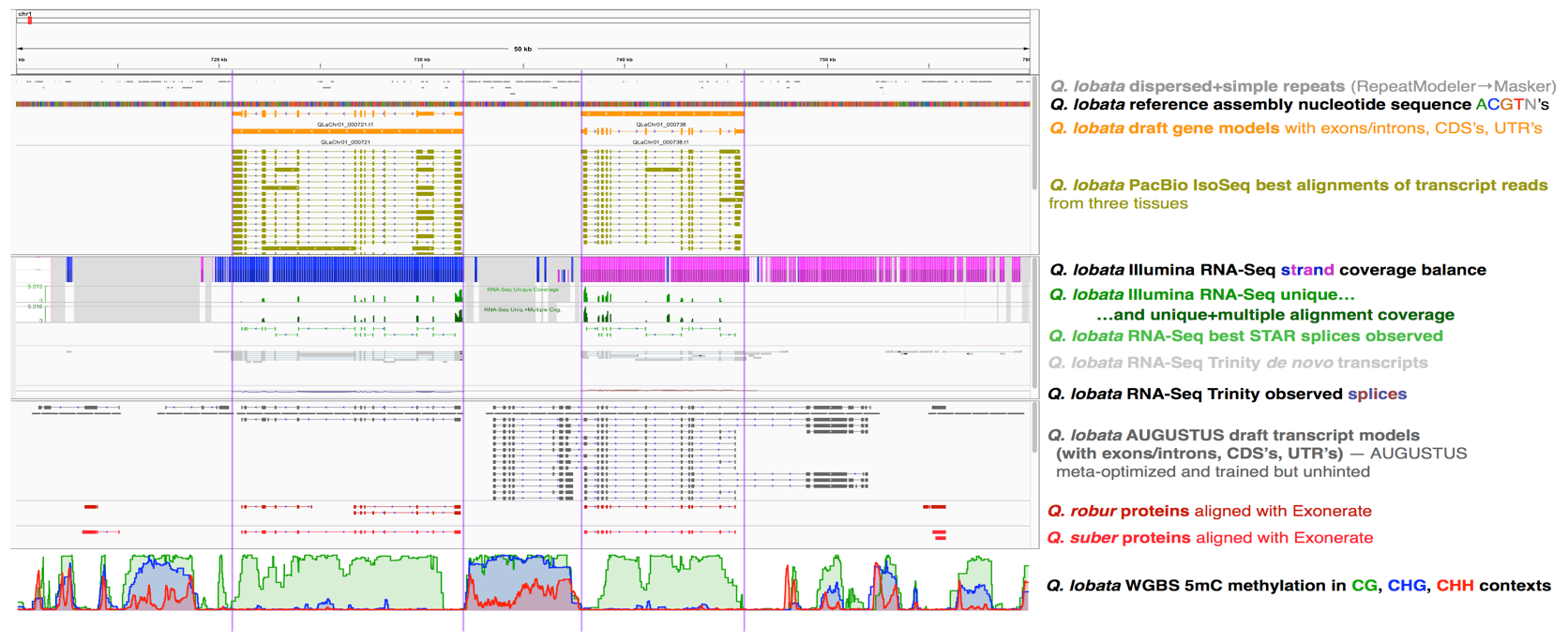
Figure 3. Snapshot of 50 Kbp (0.71 to 0.76 Mbp) from chromosome 1 of Q. lobata showing various genomic resources: dispersed and simple repeats, many tracks related to ongoing gene modeling (current consensus transcript models, PacBio long Iso-Seq transcripts, Illumina short RNA-Seq reads, Trinity-assembled RNA-Seq transcripts, AUGUSTUS predictions, aligned Q. robur and Q. suber proteins), and 5mC DNA cytosine methylation in one of our tissues (buds).